Hi-C is a great tool for genome scaffolding, but its success depends crucially on the starting assembly. It is therefore important to ensure that your starting assembly is of sufficient quality for the scaffolding task.
These recommendations cover the most common case of using Hi-C to scaffold a haploid genome assembly. Diploid or polyploid Hi-C scaffolding is possible but more complex. If you want a diploid or polyploid assembly please email support@phasegenomics.com to learn more about our recommendations.
1 Assembly technologies
Recommendations on sequencing technology for initial (contig-level) assembly
The following molecular technologies are strongly encouraged for generating starting contigs or scaffolds:
- PacBio long reads (ideally >70X coverage; minimum 30X)
- Nanopore long reads (minimum 50X; recommend error correction, less well investigated than PacBio)
The following sequencing technologies are acceptable in some cases for generating starting contigs or scaffolds:
- 10X chromium / linked-read sequencing
- Sanger / dideoxy sequencing (high coverage)
- Illumina short reads (very high coverage)
The following sequencing technologies are discouraged for generating starting contigs or scaffolds:
- Mate-pair sequencing
Why these technologies?
Long read assemblies are strongly encouraged, as they lead to extremely contiguous assemblies due to their ability to completely read through long repetitive sequences. Their relatively low accuracy and high frequency of small indels makes it desirable to perform polishing and error correction for finished genomes, but Hi-C scaffolding frequently works well even with unpolished contigs.
We recommend long-read technologies for any genome with non-trivial repeat content.
Short read / 10X-based assemblies can be “good enough” for genomes with low repeat content. Short read assemblies tend to collapse repetitive sequences and yield low contiguity assemblies with large numbers of contigs. These assemblies don’t always give good results due to their high complexity. 10X scaffolds can be ok, but other scaffolds made from short reads generally contain unacceptable levels of chimerism.
Mate pair assemblies perform poorly. While we are still accepting mate pair assemblies at the time of writing, they usually do not yield good results. Mate pair scaffolding joins short-read contigs together with long-range inserts. Unfortunately, this process is error-prone and leads to frequent misjoins, leading to extensive chimerism in the resulting scaffolds. Hi-C performs badly when there are high levels of chimerism in the starting assembly (see below). If we are given an assembly scaffolded with mate-pairs, we tend to simply break it back into contigs and try to treat it as a short-read contig assembly.
Assembly software
Recommended long-read assemblers:
- Canu
- Falcon / Falcon-Unzip (see PacBio resources for more information)
- to be continued…
2 Useful complementary data types
If available or within your budget, we strongly recommend using complementary data types to correct or improve your assembly:
- Genetic maps
- BioNano optical maps
- 10X (linked-reads)
Genetic maps contain linkage information between contigs, based on the co-segregation of specific markers on those contigs. Genetic maps can be directly used in Proximo scaffolding to constrain placement of contigs in chromosomes.
Optical mapping or linked-reads can be used to correct errors in the assembly and to perform initial scaffolding.
The best final results of Hi-C scaffolding tend to be obtained when an assembly has been previously improved using complementary technologies (e.g. PacBio + optical mapping or PacBio + genetic maps). This is particularly true for complex genomes with high repeat content.
3 Common problems in assemblies
Low contiguity
Low contiguity assemblies tend to have very large numbers of contigs. This greatly increases the complexity of the scaffolding problem, and the opportunities to make mistakes. The best way to avoid the problem of low contiguity is to use high-quality starting data (e.g. PacBio reads) at high coverage.
Contiguity is normally measured using N50, which is a statistic summarizing the size distribution of sequences. N50 is the minimum length of the set of the longest contigs that contain half the genome.
All else being equal, you want your N50 to be as large as possible. The N50 of the assembly will however depend a lot on the genome size and chromosome sizes of the species, and it is important not to push so hard for high N50 that misjoins become more common. For example, we like having N50 > 1Mbp for large plant or animal genome assemblies, but N50 = 1Mbp would be very suspicious for a brewer’s yeast (Saccharomyces cerevisiae) assembly, as most of the chromosomes of this organism are <1Mbp in length and the total genome size is only 12Mbp.
For a standard large eukaryotic genome (~1Gbp), we observe that Hi-C scaffolding usually works well if the assembly is low in errors and the starting N50 is 1Mbp or more. For this reason, we recommend that customers desiring high-quality chromosome-scale scaffolds should aim for this N50. While it is possible to go substantially lower (we have successfully scaffolded large assemblies with N50 ~ 50Kbp), results are much less reliable.
While high contiguity is desirable, it is dangerous to increase contiguity by introducing low-confidence joins of contigs. For Hi-C scaffolding we will always prefer a less contiguous assembly with high-confidence scaffolds to a more contiguous assembly with low-confidence scaffolds (see next section). However it is difficult to directly express this tradeoff numerically.
Chimeric contigs/scaffolds
Erroneously joined sequences in the starting assembly create problems in scaffolding, as different pieces of the same contig or scaffold provide refractory signals. Therefore, this fraction should be as low as possible. The best way to avoid chimeric sequences is to use high-quality initial assembly data (e.g. PacBio reads) at high coverage and to be stringent in scaffolding.
Low levels of chimerism can be accommodated and corrected using Hi-C data. As an example of chimerism in a Hi-C contact map see Figure 1, in which three large chimeric contigs are in the selection (black boxes represent selected contigs). Chimeric contigs contain distinct “squares” of contacts that in turn do not interact with each other.
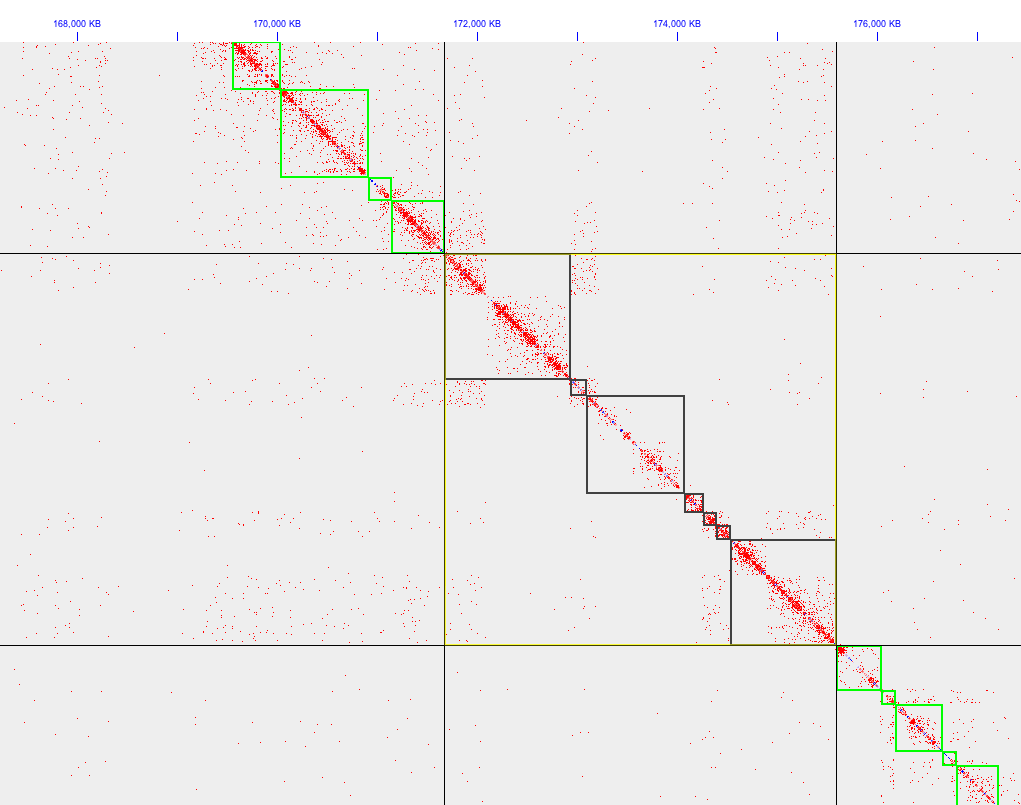
Figure 1. Three large chimeric contigs selected.
Heterozygosity
If there is heterozygosity in the starting assembly, this will lead to additional sequences being present that are homologous to some other sequence in the assembly. This can be ok if this is intentional (e.g. it is a diploid assembly or contains homeologs), but if the assembly is intended to be a single haploid sequence it will not represent linear sequence well and may contain spurious repeats. In each of these cases, Hi-C reads will be relatively difficult to map uniquely and therefore the signal useable for scaffolding will be weaker overall than for a haploid assembly. Homology is often visible as diagonal lines off the main diagonal of the Hi-C heatmap.
For an example of what homologous sequences look like on a Hi-C contact map, see Figure 2. This example shows how complex the situation can get in a repeat-rich genome, in that the homology is observed both within and between contigs and either there are structural variations between the homologs or the assembler was not properly able to assemble these contigs.
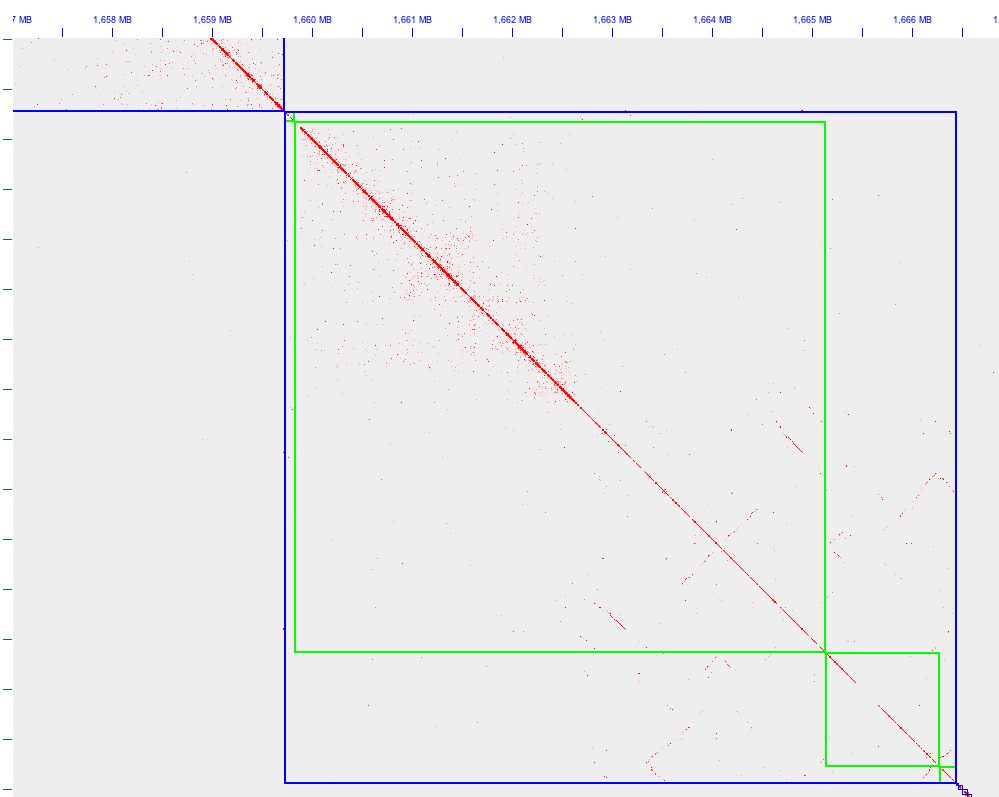
Figure 2. Homology within and between contigs.
4 Fixing problems in assemblies
We have various options for trying to fix up an assembly with the above issues.
Break contigs –> fixes chimeric contigs
Split on gaps
One is to simply split every sequence on gaps in the assembly, where a gap is defined as a stretch of Ns of at least some length (2 or 10 usually).
Polar_star
If you are using PacBio or Nanopore reads, another method is to use our tool polar_star. This tool takes your PacBio subreads (or in principle ONT reads), aligns them to your assembly, and breaks the assembly in places where long read coverage fluctuates.
Manual contig breaking
If you have aligned Hi-C data, you can use Juicebox or a similar tool to manually break contigs and make a new assembly.
Manual polishing
We will frequently perform manual fixes of an assembly using the Juicebox Assembly Tool (paper) for visualizing Hi-C data aligned to an assembly. This visualizer and interactive assembly editor is a great way to fix the last 1% of problems that benefit from human eyes after Proximo has done 99% of the work. You can move contigs, invert contigs, and break contigs in this tool. Remaking an assembly FASTA from Juicebox fixes is a little hard and not automatic, but we are working on tools that allow us to do this.
Remove homologous sequences
There are various tools to remove haplotigs, e.g. purge-haplotigs.